
| داء كانافان | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الغدد الصم، وطب الجهاز العصبي |
| من أنواع | حثل المادة البيضاء، ومرض الجهاز العصبي الوراثي التنكسي ، واضطراب صبغي جسدي متنحي ، ومرض معين |
| التاريخ | |
| سُمي باسم | ماي مور |
|
تعديل مصدري - تعديل | |
داء كانافان (بالإنجليزية: Canavan disease) هو مرض صبغي جسدي متنحي من الأمراض التنكسية التي تسبب ضموراَ يُتلف الخلايا العصبية في الدماغ. يعتبر أيضا واحد من الأمراض الدماغية التنكسية الشائعة التي تصيب الرضع.ينتج هذا المرض نتيجة النقص في انزيم امينو اسيلاز2 وهو واحد من مجموعة من الأمراض الوراثية التي يشار إليها بمرض حتل المادة البيضاء.يمتاز هذا المرض بانتكاس الميالين في الطبقة الشحمية الفسفورية التي تعزل محوار العصبون، ويرتبط هذا المرض بجين موجود في صبغ 17 في جسم الإنسان.
معدل انتشار المرض
بالرغم من أن مرض كانافان قد يظهر في أي عرق إلا أنه يصيب السلالة اليهودية في شرق أوروبا كثيراَ، فيصل عدد الأشخاص الحاملين للمرض من يهود أشكناز لحوالي شخص في كل أربعين شخص (2.5 %).
الفيزيولوجيا المرضية
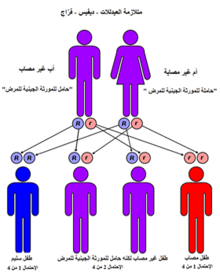
هو مرض موروث في هيئة أصباغ جسدية متنحية، فعندما يكون الوالدان حملان للمرض فإن احتمال اصابة الأبناء به تكون بنسبة 25%. توصى الأسر التي يكون فيها الأبوين حاملين للمرض بعمل اختبار للوراثة والبحث عن التوعية الوراثية. ينتج مرض كانافان عن خلل في جين اسيلاز الاسبارتات المسؤول عن إنتاج انزيم اسيلاز الاسبارتات. يمنع نقص نشاط انزيم اسيلاز الاسبارتات تكسر انزيم ن-أسيتيل الأسبارتات، حيث أن تراكم انزيم ن-أسيتيل الأسبارتات أو القصور في عملية الأيض يحول بين نمو الغمد المياليني للألياف العصبية في الدماغ.يعتبر الغمد المياليني غطاء دهني يحيط بالخلايا العصبية يعمل كعازل يسمح بالنقل الفعال للتدفع العصبي.
الأعراض
تظهر أعراض هذا المرض في مرحلة الرضاعة المبكرة وتتطور سريعاَ، وتشمل هذه الأعراض على تخلف عقلية وفقدان المهارات الحركية المكتسبة سابقاَ وصعوبات في تناول الطعام وانقباض غير طبيعي في العضلات سواء (التصلب أو الضعف) وضعف السيطرة على الرأس وتضخم الدماغ (تضخم الرأس بشكل غير طبيعي)، وقد يصاب أيضاَ بالشلل والعمى أو بالنوبات.
العلاج
لا يوجد علاج لمرض كانافان، وليس هناك مساق معياري للعلاج، ويعد العلاج لهذا المرض معالجة داعمة ومعالجة للأعراض.هناك أيضا علاج تجريبي باستخدام سيترات الليثيوم، فعندما يكون الشخص مصاب بهذا المرض فإن مستوى انزيم ن-أسيتيل الأسبارتات يكون مرتفع بشكل مزمن.ثبت في تجربة لنموذج وراثي لفأر مصاب بهذا المرض أن سيترات الليثيوم قادرة على خفض مستويات انزيم ن-أسيتيل الأسبارتات بشكل ملحوظ.وعندما تم اختبار ذلك على الإنسان انتكست حالته خلال أسبوعان من فترة الغسيل بعد سحب اليثيوم. كشف الفحص انخفاضاَ في كلِ من مستوى انزيم ن-أسيتيل الأسبارتات في المناطق المختبرة والقيم المطيافية للرنين المغناطيسي في الدماغ، والتي تعتبر أكثر سمة من سمات التطور الطبيعي وتكوًن المَيَالين. ويشير هذا الدليل أن المراقبة الكبيرة لتجربة الليثيوم قد يكون مسموح به كعلاج داعم للأطفال المصابين بمرض كانافان. بالإضافة إلى أن هناك محاولات تجريبية للمعالجة الجينية نشرت في عام 2002 م.تقتضي تلك التجارب على استخدام جين سليم للسيطرة على الجين المعيب الذي يسبب مرض كانافان. ونشرت النتائج التي تتعلق بالتجارب على الإنسان في عام 2012 م، أن هذه الطريقة ظهرت لتحسين حياة المريض بدون آثار عكسية على المريض طوال الخمسة أعوام المتتالية.
التشخيص
يؤدي هذا المرض عادة إلى الوفاة قبل سن الرابعة، ولكن قد ينجو بعض الأطفال المصابين بنوعه الخفيف حتى يبلغوا سن المراهقة والعشرينات. البحوث الحالية: كان البحث الذي اشتمل على إضافة ثلاثي الأستين في نموذج الفأر بشارة خير.يمكن لثلاثي الأستين أن ينشطر انزيمياَ ليشكل الأسيتات التي تدخل للدماغ بسرعة أكبر من الأسيتات المشحونة السالبة. يحول انزيم أَسيلاز الأسبارتات المعيب في مرض كانافان انزيم ن-أسيتيل الأسبارتات إلى أَسْبارْتات واسيتات. تمنع الطفرات التي تحدث في جين أَسيلاز الأسبارتات تكسر انزيم ن- أسيتيل الأسبارتات وتقلل من توافر اسيتات الدماغ خلال فترة نمو الدماغ، وتهدف إضافة الأسيتات باستخدام ثلاثي الأستين إلى توفير الأسيتات المفقوده ليستمر نمو الدماغ طبيعياَ. هناك فريق من الباحثين برئاسة باولا ليون حاليا في جامعة نيوجرسي للطب وطب الأسنان في مدينة ستراتفورد في ولاية نيوجرسي، ويجرى علاج جيني للدماغ في مستشفى جامعة كوبر، وتشمل الطريقة على إدخال ستة قثاطير في الدماغ تقوم بنقل محلول يحتوي على 600 مليار إلى 900 مليار من جزيئات الفيروسات التي تم إعدادها.وتم أعداد هذا الفيروس والذي يعد نسخة معدلة من الفيروس المرتبط بالفيروس الغدي ليحل محل انزيم اسيلاز الأسبارتات، وأظهرت هذه الطريقة المستخدمة في علاج الأطفال تحسنا ملحوظا في نمو الميالين وانخفاض مستويات انزيم ن-أسيتيل الأسبارتات الذيفاني.
تاريخ المرض
وصف مرتيل كانافان مرض كانافان للمرة الأولى في عام 1931 م. معهد بحوث مستشفى ميامي للأطفال غرينبرق.ف
المقال الرئيسي: معهد بحوث مستشفى ميامي للأطفال غرينبرق.ف
أحدث اكتشاف الجين المسؤول عن مرض كانافان وما تلاه من أحداث جدلاَ كبيراَ، ففي عام 1987 م قامت عائلة غرينبرق التي لديها طفلين مصابين بمرض كانافان بإعطاء عينات من الأنسجة لروبن ماتلون. كان روبن ماتلون باحثاَ في جامعة شيكاغو يبحث عن جين مرض كانافان. حدد روبن الجين المسؤول عن المرض في عام 1993م وطور فحصاَ يمكٍن الزوجين من الاستشارة قبل الإنجاب لتجنب انجاب طفل مصاب بهذا المرض. قدمت مؤسسة كانافان فحص جيني مجاني مع الفحص لفترة محددة، ولكن في عام 1997م وبعد انتقال المؤسسة إلى فلوريدا سجل موظف يعمل لدى ماتلون ومستشفى ميامي للأطفال براءة اختراع في الجين مدعياَ ملكيته للفحص الجيني مما أجبر مؤسسة كانافان على سحب اختباراتهم.تم حسم الدعوى القضائية التي رفعتها مؤسسة كانافان لاحقا ضد مستشفى ميامي للأطفال بتسوية أغلقت خارج المحكمة. يتم التطرق أحيانا إلى هذه القضية في نقاشات حول ما إذا كان تسجيل براءة الاختراع للجين يعد صواباَ.