
| مرض السُماك المرتبط بX | |
|---|---|
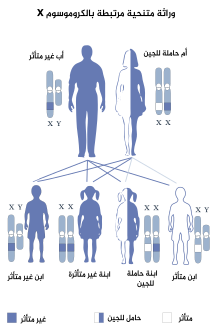 وراثة متنحية مرتبطة بX: الصبية المصابون يمكن أن يرثوا حذف أو طفرة في جين "STS” من الأم.
| |
| تسميات أخرى | Steroid sulfatase deficiency, X-linked recessive ichthyosis |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | مرض متنح مرتبط بالجنس ، وسماك |
|
تعديل مصدري - تعديل | |
مرض السُماك المرتبط ب X (بالإنجليزية: X-linked ichthyosis) وباختصار (XLI) هو حالة جلدية من أنواع السماك، ناجمة عن نقص وراثي في إنزيم سلفاتاز الستيرويد (STS) الذي يصيب 1 من كل 2000 إلى 1 من كل 6000 من الذكور. يتسبب المرض بظهور جلد جاف ومتقشر، ويرجع ذلك إلى عمليات الحذف أو طفرات في جين STS. يمكن أن يحدث المرض أيضًا في سياق عمليات الحذف الأكبر التي تسبب متلازمات الجينات المجاورة. يهدف العلاج إلى حد كبير إلى تخفيف أعراض الجلد. المصطلح مأخوذ من «الإكثي» اليونانية القديمة التي تعني «السمك».
العلامات والأعراض
تشمل الأعراض الرئيسية للـ XLI تقشر الجلد، خاصةً على الرقبة والجذع والأطراف السفلية. الأسطح الباسطة هي عادة أكثر المناطق تضرراً. يمكن أن تكون التقشرات ذات قطر > 4 مم بنية داكنة أو رمادية اللون. قد تهدأ الأعراض خلال فصل الصيف.
بعيداً عن تقشر الجلد، لا يرتبط XLI عادة مع أي مشاكل طبية رئيسية أخرى. قد يؤثر الرجفان الأذيني أو الرفرفة الأذينية على ما يصل إلى 1 من كل 10 ذكور مصابين بالـ XLI. قد تكون عتامة القرنية موجودة ولكنها لا تؤثر على الرؤية. يتم الإبلاغ عن خصية هاجرة في بعض الحالات. يمكن أيضًا اعتبار بعض الأفراد لديهم تخلف عقلي، ويُعتقد أن هذا يرجع إلى عمليات حذف تشمل الجينات المجاورة بالإضافة إلى إنزيم الSTS.
لا تعاني الإناث الحاملات للاضطراب عمومًا من أي من هذه المشاكل، ولكن نادرًا ما يصعب عليهن أثناء الولادة، لأن إنزيم ال STS المعبر عنه في المشيمة يلعب دورًا في المخاض الطبيعي. لهذا السبب يجب على الحاملين التأكد من أن طبيب التوليد على دراية بهذه الحالة.



علم الوراثة
يقع جين STS على كروموسوم X في الشريط Xp22.3. وبالتالي، فإن هذا المرض هو حالة مرتبطة بX، وكذلك فهو يؤثر على الذكور والإناث بشكل مختلف. عادةً ما يطلق على الزوج 23 من الكروموسومات «الكروموسومات الجنسية». للإناث كروموسومان X وللذكور كروموسوم X واحد والكروموسوم الآخر هو Y. لذلك، لدى الأفراد العاديين، يحمل الذكور نسخة واحدة من الجين STS والإناث تحمل نسختين. يتمكن هذا الجين من النفاذ جزئياً من تعطيل X- وعادة ما تعبر الإناث عن كميات أعلى من إنزيم STS مقارنةً بالذكور.
يمكن أن يحدث XLI من خلال عمليات الحذف أو الطفرات الجديدة في جين STS، لكن تكون وراثته أكثر شيوعًا من الأم الحاملة للمرض. يؤدي الحذف الفرداني الزيجوت أو طفرة جين STS في الذكور إلى الغياب التام لنشاط الإنزيم، في حين أن الإناث الحاملة للطفرة أو الحذف تكون مغايرة الزيجوت ولا يزال لديها نسخة طبيعية من جين STS. لا تزال الإناث الحاملات لحذف أو طفرة STS تعبر عن إنزيم STS، على الرغم من انخفاض نشاط الإنزيم.
لهذا السبب، يؤثر XLI بشكل شائع على الذكور، على الرغم من أن الأفراد الذين يعانون من تشوهات رقمية لكروموسومات الجنس (45 و X و 47 و XXY) والذين يحملون أيضًا عمليات حذف STS أو حدوث طفرات سيكونون حالات استثنائية لهذه القاعدة.
بالإضافة إلى ذلك، يمكن أن تتأثر الأنثى إذا كانت الأب مصاب بالاضطراب والأم حاملة له، حيث أنها ترث حذف أو طفرة في جين STS على كل من كروموسومات X.
نظرًا لأن غالبية الحالات تحدث من خلال توارث الحذف STS من الأم الحاملة له، يجب إجراء اختبار الإنزيم أو اختبار الحمض النووي في حالة أي حالة بسيطة تم تشخيصها حديثًا (مثل الحالة الأولى في عائلة). في حالة وجود عائلة بها العديد من الأفراد المتأثرين بالاضطراب، يمكن تحديد ما إذا كان الشخص مصاباً بالاضطراب استنادًا إلى تحليل النسب.
- إذا كان الأب متأثر بالمرض بشكل ظاهري ولكن الأم غير حاملة له (لا جينياً ولا ظاهرياً)، فعند إذن لن يتأثر الابن مطلقاً، لأنه لا يرث كرموسوم X من الأب. ولكن في نفس هذه الحالة، ستكون الابنة حاملة للمرض جينياً بلا شك.
- في حال كان الأب غير متأثر بالمرض والأم حاملة له (أي من غير أن تكون الأم مصابة به بشكل ظاهري)، فإن فرصة إصابة الابن تكون 50٪. في المقابل، تكون فرصة إصابة الابنة بالمرض بشكل ظاهري (في نفس الحالة المذكورة سابقاً) 0%. ولكن تكون فرصة إصابتها بشكل جيني 50%.
بسبب الفصل العشوائي للكروموسومات أثناء تكون الجاميتات، سيخضع كل حمل لنفس الاحتمالات، بغض النظر عن عدد النسل المتأثر سابقًا أو غير المتأثر.
تستند مخاطر تكرار الحمل، الآنفة الذكر أعلاه، إلى افتراض أن الأنثى المصابة أو الحاملة المتأثرة ستنجب أطفالًا من بينهم فرد غير متأثر أو غير حامل للمرض. من المحتمل أن تزداد مخاطر التعرض للنسل بشكل واضح في حالة وجود اتحاد بين ذكر مصاب بـ XLI وامرأة حاملة له.
التشخيص
يمكن أن يُشتبه بال XLI على أساس النتائج السريرية، على الرغم من أن الأعراض قد تستغرق فترات متفاوتة لتصبح واضحة، على سبيل المثال من بضع ساعات بعد الولادة، وحتى سنة في الحالات الأكثر اعتدالاً. يتم التشخيص عادة من قبل طبيب الأمراض الجلدية، الذي ينظم خطة العلاج.
يتم تأكيد نقص إنزيم STS باستخدام فحص كيميائي حيوي متاح سريريًا. يمكن إجراء الكشف عن الحاملين للاضطراب في أمهات الأبناء المصابين باستخدام هذا الاختبار. كما يتم تقديم اختبار جزيئي لحذف DNA أو طفراته، ويمكن أن يكون ذلك مفيدًا بشكل خاص في تقييم الأفراد الذين يعانون من حالات طبية مرتبطة. يمكن إجراء اختبار ما قبل الولادة باستخدام الاختبارات الكيميائية أو الجزيئية. ومع ذلك، فإن استخدام تشخيص ما قبل الولادة للحالات الوراثية التي تعتبر عمومًا حميدة يثير اعتبارات أخلاقية خطيرة ويتطلب استشارات وراثية مفصلة.
العلاج
لأن XLI ناتج عن طفرة جينية أو حذفها، فلا يوجد «علاج». أحد أهداف العلاجات الحالية هو تقليل التقشر عن طريق إزالة المقاييس الزائدة والقشارية والحفاظ على رطوبة البشرة. يمكن تحقيق ذلك باستخدام مجموعة متنوعة من الكريمات الموضعية
- تستخدم العوامل الكيراتينية مثل لاكتات الأمونيوم (Lac-Hydrin) لتسهيل إطلاق الخلايا القرنية المحفوظة.
- آيزوتريتينوين الموضعي.
البحث لا يزال مستمر فيما يتعلق باستخدام العلاج الجيني من أجل علاج XLI.
تاريخ
في الستينيات من القرن الماضي، تم تمييز مرض السماك المتنحي المرتبط بX سريريًا عن السماكات الأخرى.
انظر أيضًا
مراجع
روابط خارجية
| تصنيف | |
|---|---|
| موارد خارجية |