
| داء ولمان | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
|
تعديل مصدري - تعديل | |
داء ولمان (بالإنجليزية: Wolman disease) والمعروف أيضا ببداية ظهور نقص في الليباز الحمضي في الجسيمات الحالة وهو مرض وراثي نادر يسببه نقص إنزيم يعرف باسم الليباز الحمضي في الجسيمات الحالة. ويعد هذا الإنزيم ضروري لتحليل بعض الدهون داخل الخلايا، ويؤدي نقصه إلى تراكم الدهون في الكبد والأمعاء وأجزاء أخرى من الجسم. ينتمي مرض ولمان إلى مجموعة من الأمراض المعروفة باسم أمراض الاختزان في الجسميات الحالة. وظيفة الجسيمات الحالة -بصفتها مراكز إعادة التدوير داخل الخلايا- هي تحويل عدد من المواد غير المرغوب فيها إلى مواد يمكن للخلية إعادة استخدامها.الإنزيمات هي بروتينات عالية التخصص داخل الجسيمات الحالة والتي تحلل مواد غذائية معينة أو تهضمها مثل بعض الدهون والكربوهيدرات. عندما تكون هذه الأنزيمات معيبة أو عندما لا تتواجد تماماً بسبب طفره وراثية، ينشئ هذا الإنزيم نتيجة للتراكم غير الطبيعي للمواد في خلايا الجسم. ويعد مرض ولمان الشكل المبكر لبداية نقص LAL. يظهر المرض بشكل متقدم عادة خلال الأسابيع أو الأشهر الأولى من الحياة بينما يظهر الشكل المتأخر والذي يعرف بمرض اختزان كوليسترول استر (CESD) عادة في وقت متأخر من مرحلة الطفولة أو حتى مرحلة البلوغ.
التشخيص
ما هي مسببات داء ولمان؟
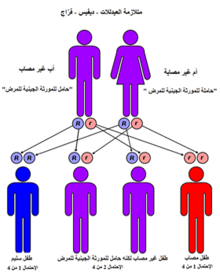
لمرض ولمان نمط وراثي جسمي مقهور ففي كل شخص نسختين من جين LAL (التي تعرف أحيانا باسم جين LIPA) نسخة متوارثة من الأب والآخرى من الأم ويحدث نقص LAL عندما يكون لدى الشخص طفرة في نسختي جين LAL. ويحمل كلاً والدا المريض الذي يعاني من نقص LAL نسخة واحدة معيبه من جين LAL. مع كل حمل احتمالية إنجاب طفل مصاب آخر هي %25 لمن لديهم ابن أو ابنه مصاب بنقص LAL. كل شخص ولد بعيوب خلقية في كلا جينيّ LAL غير قادر على إنتاج كميات كافية من الإنزيم LAL.
تعمل الجسيمات الحالة كمراكز إعادة تدوير داخل الخلايا والتي بوظيفتها تقوم بالتخلص من المواد غير المرغوب فيها وتحويلها إلى مواد يمكن إعادة استخدامها والاستفادة منها. ويعمل الإنزيم LAL على توزيع دهون معينه داخل الجسيمات الحالة. عندما لا يكون في الجسيمات الحالة ما يكفي من انزيم LAL فإنها لن تكون قادره على التخلص من هذه الدهون.
ماهي نسبة شيوع داء ولمان؟
مرض ولمان كأي مرض نادر آخر، حيث لا يشخص نهائياً أو قد يخطأ في تشخصيه، مما يعني أنه ليس هناك الكثير من المعلومات الطبية حول مدى شيوع هذه الحالة. ويصيب هذا المرض الذكور والإناث بأعداد متساوية. اكتشفت أحد التقارير 8 حالات مصابة بمرض ولمان من بين أكثر من 4.2 مليون حالة ولادة خلال فترة 16 عاماً. يفسر هذا إصابة حوالي شخص واحد من بين 500,000 حالة ولادة ويعني هذا بالنسبة لبلد كالولايات المتحدة، انه قد يولد 8 أطفال مصابين بمرض ولمان كل عام.
هل يصيب داء ولمان فئات معينة من السكان بشكل متكرر أكثر من غيرهم؟
من المتوقع أن يصيب فئات معينة من السكان بشكل أكبر لأن لمرض ولمان نمط وراثي، حيث يكون هنالك تزواج بين الأفراد ذو صلة وراثياً (زواج الأقارب). فقد ولِد العديد من المصابين بجين LAL غير الطبيعي مؤخراً من السكان الإيرانيين اليهوديين.
يشتبه تشخيص داء ولمان في الأطفال حديثي الولادة بناء على عدة أعراض مثل تضخم الكبد غير الطبيعي ومشاكل في الجهاز الهضمي. يمكن تأكيد التشخيص عن طريق الفحص السريري الدقيق وعن طريق التاريخ المفصل للمريض (بما في ذلك التاريخ العائلي) والاختبارات المتخصصة التي تكشف عن عدم وجود أو نقص النشاط لليباز إنزيم حمض الليزوزومية (LAL) في بعض خلايا وأنسجة الجسم. يمكن أن يتم التشخيص أيضا عن طريق التأكد من وجود طفرات (عيوب) في نسختي الجين LIPA. ومن الممكن التشخيص قبل الولادة من خلال أخذ عينات من الزغابات المشيمائية (CVS) أو بزل السلى. تأخذ عينات من أنسجة الجنين خلال أخذ عينات من الزغابات المشيمائية CVS ويتم إجراء اختبارات إنزيم (المقايسات) على خلايا الأنسجة المستزرعة (الليفية) أو خلايا الدم البيضاء (الكريات البيضاء). اما في حالة إجراء اختبار بزل السلى فيؤخذ عينة من السائل الذي يحيط بالجنين وتتم دراستة.
الأعراض
تظهر مؤشرات وأعراض داء ولمان عادة بعد مده قصيرة من الولادة وعادة في الأسابيع القليلة الأولى من حياة الرضيع. وقد يظهر على الرضع المصابين الأعراض الآتية:
- - صعوبة في التغذية مع التقيء المتكرر.
- - الإسهال (التبرز بشكل متكرر).
- - تورم في البطن (انتفاخ البطن).
- - تضخم الكبد والطحال (تضخم الطحال).
- - صعوبة في كسب الوزن أو فقدان الوزن في بعض الأحيان.
يؤدي تراكم الدهون في جدران الأمعاء إلى مشاكل خطيرة في الجهاز الهضمي وسوء الامتصاص الذي يؤدي إلى عدم قدرة الأمعاء على امتصاص العناصر الغذائية والسعرات الحرارية من الطعام. ويرتبط سوء الامتصاص غالباً بالتقيؤ المستمر والإسهال المتكرر وظهور براز دهني كريه الرائحة (إسهال دهني). وبسبب ظهور هذه المضاعفات في الجهاز الهضمي، عادة ما يصاب الرضع المصابين بالمرض بنقص في النمو وعدم القدرة على اكتساب الوزن المناسب لسنهم (فشل نمو).
ويؤدي تقدم المرض وازدياد تراكم الدهون في الكبد إلى مضاعفات أخرى والتي قد تشمل اصفرار الجلد وبياض العينين (اليرقان)، وحمى انخفاض درجة الحرارة.
من النتائج المرتبطة بأمراض ولمان هو تراكم المواد الطباشيرية (تكلس) في الغدة الكظرية. تقع الغدد الكظرية فوق الكلى وتعمل على إنتاج الهرمونات (وهي المواد الكيميائية التي تساعد على تنظيم وظائف مختلفة في الجسم). لا يمكن الكشف عن تكلس الغدد الكظرية بواسطة الفحص البدني، ولكن يمكن أن يكشف عنه من خلال الأشعة السينية أو الأشعة المقطعية أو طرق تصوير أخرى. وتعد هذه الوسائل مفيدة جداً في المساعدة على جعل التشخيص أكثر دقة، كما أن هناك عدد قليل جداً من الحالات الأخرى التي تسبب التقيؤ، وعدم القدرة على اكتساب الوزن وتكلس الغدد الكظرية. لا تٌظهر جميع الحالات المصابة بمرض ولمان هذه الأعراض عند التشخيص، فهذه الاعراض تظهر في الحالات الأشد خطورة، وغالبا مايتأخر ظهورها. وقد يمنع تكلس الغدد الكظرية إنتاج الهرمونات الأساسية الكافية ويمكن أيضاً أن يؤثر على عملية التمثيل الغذائي وضغط الدم والجهاز المناعي والعمليات الحيوية الأخرى في الجسم. ولم يثبت بعد أن تكلس الكظرية يعيق وظيفة الغدة الكظرية.
و تتقدم مضاعفات المرض مع مرور الوقت، مما يؤدي في نهاية المطاف إلى مشاكل صحية قد تهدد الحياة مثل انخفاض مستويات خلايا الدم الحمراء في الدم (فقر الدم) وضعف أو فشل الكبد والهزال الجسدي (دنف).
العلاج
لم يتم اعتماد أي علاج لمرض ولمان إلى الآن، ولم يتم أيضاً تحديد أي علاج في التجارب السريرية للحد من التشوهات التي تصيب المرضى الذين يعانون من مرض ولمان. ويتركز العلاج بشكل رئيسي على الحد من مضاعفات معينة وقد تختلف هذه المضاعفات بين المرضى. وكثير ما يتم تقديم العلاج في مراكز متخصصة، وقد تتطلب تدخل من فريق مكون من اختصاصيين من مختلف الاختصاصات. وقد تفيد الاستشارة الوراثية الأشخاص المتضررين وأسرهم. ويمكن أن تشمل هذه التدخلات على مايلي:
- - تغيير مصدر الحليب سواء كان من الثدي أو من حليب صناعي إلى حليب مخصص قليل الدسم.
- - إعطاء مقدمة عن الإطعام الوريدي (التغذية بالحقن).
- - تزويد المريض بمضادات حيوية للالتهابات.
- - تقديم علاج الستيرويد التعويضي وذلك بسبب المخاوف المتعلقه حول تأثر وظيفة الغدة الكظرية.
وتؤكد الدراسات احتمالية وفاة معظم الأطفال المصابين بمرض ولمان قبل ولادتهم. فقد سجلت التقارير ان الناجين من مرض ولمان الأكبر سناً لعام 2009 كانت أعمارهم 4 سنوات و11 سنة وتم علاجهمها عن طريق زراعة الخلايا الجذعية للدم.
العلاج التجريبي
تم معالجة بعض الأطفال الذين يعانون من مرض ولمان علاجا تجريبياً يسمى زراعة الخلايا الجذعية للدم (HSCT)، والمعروف أيضا باسم زراعة النخاع العظمي، وذلك لمحاولة منع ازدياد سوء حالة المرض.
الخلايا الجذعية المكونة للدم هي خلايا متخصصة موجودة في نخاع العظم (مادة إسفنجية ناعمة تتواجد في العظام الطويلة). تنمو هذه الخلايا الجذعية في الدم وتتطور إلى أن تصبح أحد أنواع خلايا الدم الرئيسية الثلاثة: خلايا الدم الحمراء أو خلايا الدم البيضاء أو الصفائح الدموية.
استبدال خلايا الجذع، والذي يتطلب إبقاء الطفل في المستشفى وتزويده بأدوية قوية جداً قبل وبعد العملية، ولهذا مخاطر كبيرة وفوائد محتملة ايضاً.
- تحتوي الخلايا السليمة التي ينتجها النخاع الجديد على كميات كافية من حمض ليباز الليزوزومية واللازم للتخلص من الدهون الثلاثية والكولسترول.
- وقد ظهر على الأشخاص المصابين بمرض ولمان والذين تم معاجلتهم عن طريق زراعة الخلايا الجذعية للدم تحسن في الأعراض الحالية وتجنب ظهور مضاعفات إضافية مثل فشل الكبد.
- ترتبط زراعة الخلايا الجذعية للدم في حالة مرض وولمان في الوقت الحاضر بارتفاع نسبة حدوث مضاعفات خطيرة بما في ذلك الوفاة وداء الطعم حيال الثوي وغيرها من الآثار طويلة المدى والتي من الممكن ظهورها بعمر متأخر.
توجيهات البحوث
يدرس الباحثون حاليا علاج إنزيمي تعويضي لحالات LSDs مثل مرض ولمان. ويشمل العلاج الإنزيمي التعويضي الاستعاضة بالإنزيم المفقود في الأشخاص الذين يعانون من نقص أو انعدام إنزيم معين. وقد تم تطوير إصدارات اصطناعية للإنزيمات المفقودة وتستخدم لعلاج الأشخاص الذين يعانون من امراض LSDs أخرى بما في ذلك مرض فابري ومرض قوثر بنجاح. كما تجرى دراسة لعلاج جيني كنهج آخر محتمل لمعالجة امراض LSDs. تستبدل الجينات المعيبة الموجودة في المريض في العلاج الجيني بجين عادي لتمكين إنتاج الإنزيم النشط ومنع تنمية وتطور المرض. ونظراً للنقل الدائم من الجينات العادية القادرة على إنتاج إنزيم نشط في جميع أماكن المرض. ويعد هذا النوع من العلاج نظرياً الأكثر احتمالاً إلى “معالجة" المرض وعلى الرغم من ذلك توجد العديد من الصعوبات التقنية التي يجب حلها قبل نجاح العلاج الجيني في وفتنا الحالي.
تطلب شركة سيناجيفا بيوفارما ليكسينغتون بولاية ماساتشوستس من المرضى المشاركة في التجارب السريرية والتي تجرى لتقييم العلاج الإنزيمي التعويضي لنقص الليباز الحمضي في الجسيمات الحالة.
التسمية
سمي المرض بهذا الاسم نسبة لموشيه وولمان.